Endothelial dysfunction: cause of the insulin resistance syndrome.
Pinkney JH, Stehouwer CD, Coppack SW, Yudkin JS.
Source
Department of Medicine, University of Bristol, U.K.
Abstract
Insulin resistance has been proposed as the metabolic basis of atherogenesis. This hypothesis is based on the concept of the "insulin resistance syndrome," according to which insulin resistance is viewed as the primary abnormality that gives rise to dyslipidemia, essential hypertension, impaired glucose tolerance, and NIDDM. However, this hypothesis takes no account of the well-established and central role of vascular endothelium in the atherogenic process. Although endothelial injury is an early and prominent feature of atherogenesis, relatively little attention has been given to its metabolic consequences. In subjects with NIDDM, we have shown that endothelial dysfunction is associated with insulin resistance, raising the question of whether this relationship could be causal. In this article, we review the factors that are considered to be responsible for the development of endothelial dysfunction during atherogenesis, together with the metabolic consequences of endothelial dysfunction. While dysfunction of the endothelium in large and medium-sized arteries plays a central role in atherogenesis, we argue that dysfunction of peripheral vascular endothelium, at arteriolar and capillary level, plays the primary role in the pathogenesis of both insulin resistance and the associated features of the insulin resistance syndrome. We propose that the insulin resistance syndrome, together with many aspects of atherogenesis, can be viewed as the diverse consequences of endothelial dysfunction in different vascular beds. This new and testable hypothesis accounts for both the endothelial and metabolic abnormalities associated with atherogenesis.
Reduced expansion of the capillary network, with attenuation of microcirculatory blood flow to metabolically active tissues, contributes to the impairment of insulin-stimulated glucose and lipid metabolism.
Ουσιαστικά φαίνεται ότι η ζημιά γίνεται στα ενδοθηλιακά κύτταρα που βρίσκονται στα τοιχώματα των αιμοφόρων αγγείων και τα οποία τροφοδοτούν τους θύλακες.

Εδώ φαίνεται η διαφορά υγιή αγγείων

Εδώ με κόκκινο και μπλε χρώμα φαίνεται το δίκτυο τροφοδοσίας του θύλακα και των γύρω ιστών του δέρματος.
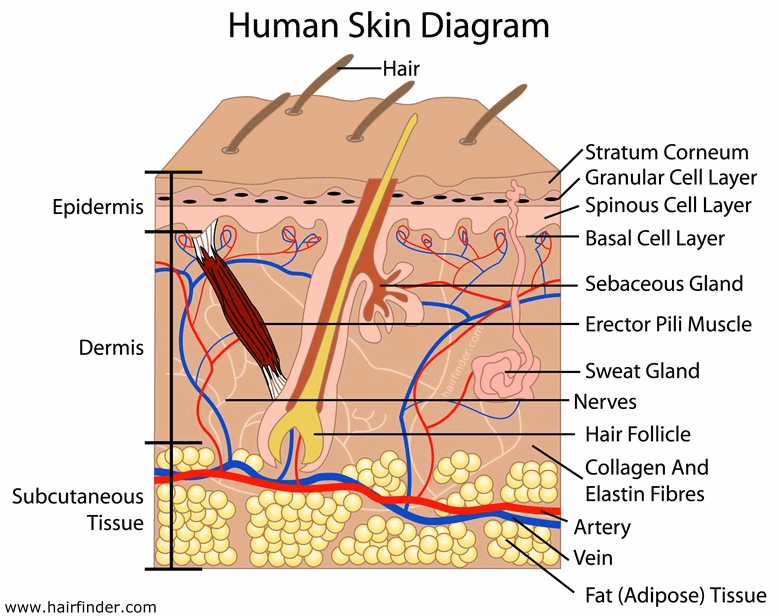
Σιγά σιγά η ροή του αίματος όλο και μειώνεται, δημιουργείται φλεγμονή στην περιοχή, υποξία και το αποτέλεσμα με τα χρόνια είναι αυτό που βλέπουμε στον καθρέπτη. Στο τελικό αποτέλεσμα που φαίνεται ένα δέρμα λεπτό, γυαλιστερό και άτριχο είναι το αποτέλεσμα μιας περιοχής που η παροχή αίματος και οξυγόνου, έχει σχεδόν εξαφανιστεί. Το ίδιο ακριβώς συμβαίνει σε διαβητικές περιοχές στο σώμα με διαβήτη τύπου 2, ο οποίος προκαλεί κάτι ανάλογο, σε μεγαλύτερο όμως βαθμό.
Η φιναστερίδη και η μινοξιδίλη σε αρχικά στάδια μπορούν να φρενάρουν αυτό το φαινόμενο με τη φιναστερίδη να διορθώνει σε κάποιο βαθμό τον μεταβολισμό της γλυκόζης, άρα και της ινσουλίνης γενικότερα, κάτι που έχει επίσης επίδραση στα αδρογόνα μέσω της SHBG που αυξάνεται όσο βελτιώνεται η αντίσταση στην ινσουλίνη και την μινοξιδίλη να προκαλεί αγγειοδιαστολή τοπικά ώστε να περάσει περισσότερο αίμα καθώς και κατά την απορρόφησή της να προκαλεί μικρότερη έκκριση ινσουλίνης από τα b cells.
Και η αντίσταση στην ινσουλίνη και το πρόβλημα της ελλιπής αιμάτωσης, είχε, έχει και θα συνεχίζει να απορρίπτεται από την επιστήμη.
Mechanisms of glycation in atherogenesis.
Pamplona R, Bellmunt MJ, Portero M, Prat J.
Source
Dpt Ciències Mèdiques Bàsiques, Faculty of Medicine, University of Lleida, Spain.
Abstract
Non-enzymatic glycation may affect the arterial wall altering its connective tissue and promoting LDL accumulation. Its recognition by specific receptors and growth factor release, as well as possible alteration of DNA, may stimulate smooth muscle cell proliferation. Free radical generation may favour non-enzymatic PUFA degradation and quench NO, which would alter vascular relaxation. All of these aspects may participate in atherogenesis.
Pinkney JH, Stehouwer CD, Coppack SW, Yudkin JS.
Source
Department of Medicine, University of Bristol, U.K.
Abstract
Insulin resistance has been proposed as the metabolic basis of atherogenesis. This hypothesis is based on the concept of the "insulin resistance syndrome," according to which insulin resistance is viewed as the primary abnormality that gives rise to dyslipidemia, essential hypertension, impaired glucose tolerance, and NIDDM. However, this hypothesis takes no account of the well-established and central role of vascular endothelium in the atherogenic process. Although endothelial injury is an early and prominent feature of atherogenesis, relatively little attention has been given to its metabolic consequences. In subjects with NIDDM, we have shown that endothelial dysfunction is associated with insulin resistance, raising the question of whether this relationship could be causal. In this article, we review the factors that are considered to be responsible for the development of endothelial dysfunction during atherogenesis, together with the metabolic consequences of endothelial dysfunction. While dysfunction of the endothelium in large and medium-sized arteries plays a central role in atherogenesis, we argue that dysfunction of peripheral vascular endothelium, at arteriolar and capillary level, plays the primary role in the pathogenesis of both insulin resistance and the associated features of the insulin resistance syndrome. We propose that the insulin resistance syndrome, together with many aspects of atherogenesis, can be viewed as the diverse consequences of endothelial dysfunction in different vascular beds. This new and testable hypothesis accounts for both the endothelial and metabolic abnormalities associated with atherogenesis.
Reduced expansion of the capillary network, with attenuation of microcirculatory blood flow to metabolically active tissues, contributes to the impairment of insulin-stimulated glucose and lipid metabolism.
Ουσιαστικά φαίνεται ότι η ζημιά γίνεται στα ενδοθηλιακά κύτταρα που βρίσκονται στα τοιχώματα των αιμοφόρων αγγείων και τα οποία τροφοδοτούν τους θύλακες.
Εδώ φαίνεται η διαφορά υγιή αγγείων
Εδώ με κόκκινο και μπλε χρώμα φαίνεται το δίκτυο τροφοδοσίας του θύλακα και των γύρω ιστών του δέρματος.
Σιγά σιγά η ροή του αίματος όλο και μειώνεται, δημιουργείται φλεγμονή στην περιοχή, υποξία και το αποτέλεσμα με τα χρόνια είναι αυτό που βλέπουμε στον καθρέπτη. Στο τελικό αποτέλεσμα που φαίνεται ένα δέρμα λεπτό, γυαλιστερό και άτριχο είναι το αποτέλεσμα μιας περιοχής που η παροχή αίματος και οξυγόνου, έχει σχεδόν εξαφανιστεί. Το ίδιο ακριβώς συμβαίνει σε διαβητικές περιοχές στο σώμα με διαβήτη τύπου 2, ο οποίος προκαλεί κάτι ανάλογο, σε μεγαλύτερο όμως βαθμό.
Η φιναστερίδη και η μινοξιδίλη σε αρχικά στάδια μπορούν να φρενάρουν αυτό το φαινόμενο με τη φιναστερίδη να διορθώνει σε κάποιο βαθμό τον μεταβολισμό της γλυκόζης, άρα και της ινσουλίνης γενικότερα, κάτι που έχει επίσης επίδραση στα αδρογόνα μέσω της SHBG που αυξάνεται όσο βελτιώνεται η αντίσταση στην ινσουλίνη και την μινοξιδίλη να προκαλεί αγγειοδιαστολή τοπικά ώστε να περάσει περισσότερο αίμα καθώς και κατά την απορρόφησή της να προκαλεί μικρότερη έκκριση ινσουλίνης από τα b cells.
Και η αντίσταση στην ινσουλίνη και το πρόβλημα της ελλιπής αιμάτωσης, είχε, έχει και θα συνεχίζει να απορρίπτεται από την επιστήμη.
Mechanisms of glycation in atherogenesis.
Pamplona R, Bellmunt MJ, Portero M, Prat J.
Source
Dpt Ciències Mèdiques Bàsiques, Faculty of Medicine, University of Lleida, Spain.
Abstract
Non-enzymatic glycation may affect the arterial wall altering its connective tissue and promoting LDL accumulation. Its recognition by specific receptors and growth factor release, as well as possible alteration of DNA, may stimulate smooth muscle cell proliferation. Free radical generation may favour non-enzymatic PUFA degradation and quench NO, which would alter vascular relaxation. All of these aspects may participate in atherogenesis.



")